Hoboken, New Jersey: March 10, 2021: Castor, a leading provider of clinical trial technology, today announced the signing of a top-5 pharmaceutical company and expansion of its global team.
2020 was a defining year for Castor. The company launched its scalable decentralized clinical trial platform and integrated product offerings that created the foundation for the company to realize 383% commercial growth. Castor now has more than 400 recurring paying customers, including a top-5 pharma company signed in Q1.
On the early success of 2021, Castor CEO Derk Arts, MD, PhD, said “Historically, our products saw the strongest traction in the mid-market, but it’s clear that all segments are in need of a scalable, self-service platform to run more patient-centric trials. We are seeing growth in the big pharma segment ahead of our own plans, which is an indication of how COVID-19 accelerated the adoption of innovation.”
Looking forward, the company plans for 2021 to become the year in which they make decentralized trials scale, getting the technology in the hands of as many users as possible. Castor has a track-record in global study deployments, having supported more than 7,500 studies on its platform. The company will continue to deploy its suite of tools to enable enrollment, screening, and consenting in a fully remote or hybrid fashion. In parallel, the company’s mobile offerings are also expanding with the imminent release of its new ePRO app for iOS and Android.
Additionally, the company plans to capitalize on their investments in data standardization, by letting their first users experiment with digital twins: synthetic patients generated from metadata available from previous projects and the actual patients in the trial.
“For the future growth of Castor, it is instrumental that we keep looking for the best talent out there. We added more than 35 new hires to our ranks in the past four months, with a combined industry experience of more than 200 years,” said Derk. Castor’s new hires include the following:
Daniel Silva joins Castor’s Management team as VP of Customer Success and brings more than 20 years of experience in building customer success organizations at Oracle, TraceLink, and endpoint Clinical. “As a young technology company grows, it is crucial to have a scalable customer success model in place that can continue to provide customers with a superior overall experience,” said Dan. “Castor’s customers rate our products and services as one of the best in the eClinical industry*. I am excited to have the opportunity to continue to provide our customers with an outstanding experience as we double the number of customers in the coming year.”
Senior Product Manager Sébastien Bohn brings more than 20 years of product management experience in the life sciences space at IBM, Merge Healthcare (acquired by IBM), and KIKA Medical International (acquired by Merge eClinical). “With Castor I see a massive opportunity to scale decentralized clinical trials for the first time, thanks to a technology-first approach, consisting of multiple native integrated product lines. This includes recruitment landing pages, enrollment, eConsent, EDC, reporting, and data analysis,” said Sébastien.
Director of Quality and Compliance Fatma Elfaghi brings more than 10 years of experience in managing compliance and regulatory affairs at Anju Software and OmniComm Systems. “It is amazing to see the foundation of Castor’s platform and the compliance procedures and documentation already in place that have resulted in successfully completed audits from major pharmaceutical companies,” said Fatma.
Castor is determined to hire more than 30 additional people in Q2 and Q3 to expand its global team.
About Castor
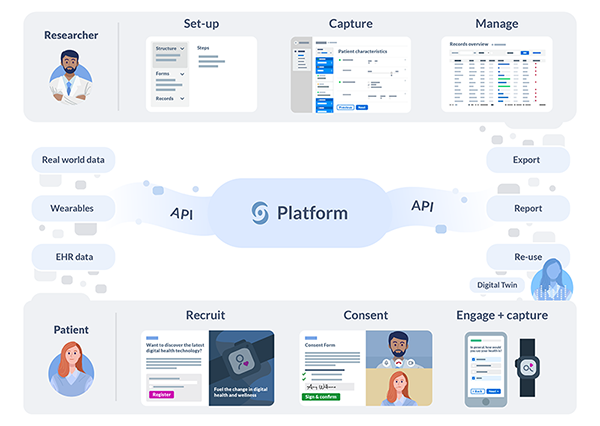
Based in the United States and The Netherlands, Castor is an international health-tech company founded by CEO Derk Arts, MD, PhD. Their cloud-based clinical data platform simplifies the clinical trial process, from recruitment to analysis, for researchers worldwide.
More than 75,000 researchers across 90 countries are using Castor to supercharge their research. Castor’s platform has supported more than 7,500 commercial and academic studies that cover a broad range of therapeutic areas including diabetes, cardiovascular disease, rare diseases, infectious diseases, and oncology. Researchers on the platform generate vast amounts of data from traditional and remote trials, and Castor recently reached milestones of 250,000,000 data points and 2,500,000 enrolled patients. Castor’s goal is to make the world’s research data reusable, enabling AI-driven clinical trials, and ultimately creating a future in which they maximize the impact of data through reuse.
In 2020, Castor raised a $12M Series A from Two Sigma Ventures with participation from Hambrecht Ducera Growth Ventures and existing investor INKEF Capital. Castor previously raised a $6.25M seed round from INKEF Capital in 2018.
LinkedIn: www.linkedin.com/company/ciwit-b-v-
Twitter: www.twitter.com/castor
*Castor rated 4.7 out of 5 stars, from more than 100 customer reviews on Capterra.


 Cherié L. Butts, PhD, Medical Director and Head of Clinical Assessments at Biogen and newly appointed Castor Advisory Board member, commented:
Cherié L. Butts, PhD, Medical Director and Head of Clinical Assessments at Biogen and newly appointed Castor Advisory Board member, commented: