As decentralized clinical trials (DCTs) gain popularity and a nod of regulatory approval—the FDA just released updated guidance in January 2021—researchers are looking to incorporate remote technologies into medical device trials for a wide range of device classifications. For example, can a Class IIb device be managed entirely from home? Can at-home monitoring be used at all for a Class III device? This article explores current regulations for various classes of medical devices and how this will impact the need for participants to visit research sites throughout the trial.
The Benefits
There are numerous ways medical device trials can benefit from DCT elements:
- Many devices can be shipped directly to patients.
- Connected devices can push data directly to the trial’s master data file through a cloud platform, reducing the need for source data verification in some cases.
- Devices that used DCT infrastructure during the investigative phase need comparatively little work to set up Post-Market Surveillance (PMS).
Understanding the relevant regulations
Medical devices are highly regulated to prevent harm to the general public. Although regulations vary from country to country, the primary concerns are invariably patient safety and privacy. When sifting through the regulatory considerations, you will most likely need to consider these key rules and agencies:
Clinical Evaluation Report (CER): This report documents the conclusions of a clinical evaluation of your medical device. It demonstrates that your device achieves its intended purpose without exposing users and patients to further risk.
General Data Protection Regulation (GDPR): A regulation in EU law on data protection and privacy in the European Union (EU) and the European Economic Area (EEA) aimed at giving individuals control over their personal data.
ISO 14155: According to ISO.org, these principles address “good clinical practice for the design, conduct, recording, and reporting of clinical investigations carried out in human subjects to assess the safety or performance of medical devices for regulatory purposes.”
EU MDR: In May 2021, The EU MDR replaced the EU’s Medical Device Directive (93/42/EEC) and Directive on Active Implantable Medical Devices (90/385/EEC). Manufacturers selling medical devices within the EU must adhere to these new regulations to ensure their products are safe to use.
FDA: The FDA requires firms that manufacture, repackage, relabel and/or import medical devices sold in the United States to comply with their basic regulatory requirements which ensure the quality and safety of products.
Conducting medical device clinical trials in Europe can be quite challenging—with or without DCT considerations—as the MDR contains approximately 300 line items. Thankfully, many match up with the ISO 14155 regulations or standards, so there’s a harmonization effort. For example, the Annex 15 of the MDR outlines protocol for pre-market clinical investigations is very much aligned with the ISO 14155.
Understanding medical device classifications
All medical devices fall under a category of the medical device classification system. The Medical Device Amendments of 1976 to the Federal Food, Drug, and Cosmetic Act established three regulatory classes for medical devices—although those classes have been broken down further since.
Classification is based on the degree of control required to assure the safety and effectiveness of the various types of devices. Typically, low-risk devices (e.g. eyeglasses) can be self-certified while high-risk devices (e.g. pacemakers) require permanent monitoring.
Class I
Class I medical devices are non-invasive devices or equipment that present minimal potential for harm to the patient or user. According to the FDA, 47% of medical devices fall under this category and 95% of these are exempted from the regulatory process. Examples include bandages, canes, or compression stockings.
- Class Is
Class Is is a sub-group of similarly non-invasive devices and includes sterile devices. Examples include examination gloves, oxygen masks, and stethoscopes. - Class Im
Class Im devices are considered low-risk and encompass measuring devices. Examples include thermometers and non-invasive blood pressure machines.
Class II
According to the FDA, 43% of medical devices fall into this category, which includes everything from home pregnancy tests to wheelchairs.
- Class IIa
Class IIa devices are low-to-medium risk devices. These may be installed within the body for a short time, between an hour to 30 days. Examples include catheters and hearing aids. - Class IIb
Class IIb devices are typically medium-to-high risk which may be installed within the body for 30 days or longer. Examples include equipment for intensive care monitoring and ventilators.
Class III
Class III devices are high-risk, implanted, or used to sustain or support life. According to the FDA, 10% of medical devices fall under the category of class III devices. Examples include orthopedic implants, pacemakers, deep brain stimulators, and prosthetic heart valves.
Using remote technology in a medical device trial
Almost any medical device study protocol can incorporate elements of DCT. Although some trials may require a clinician to administer or operate the device, almost all device trials can still leverage remote technology. Here’s how:
Televisits
Televisits can eliminate the need for on-site clinical assessments in many medical device trials through video interaction with patients and online assessments,
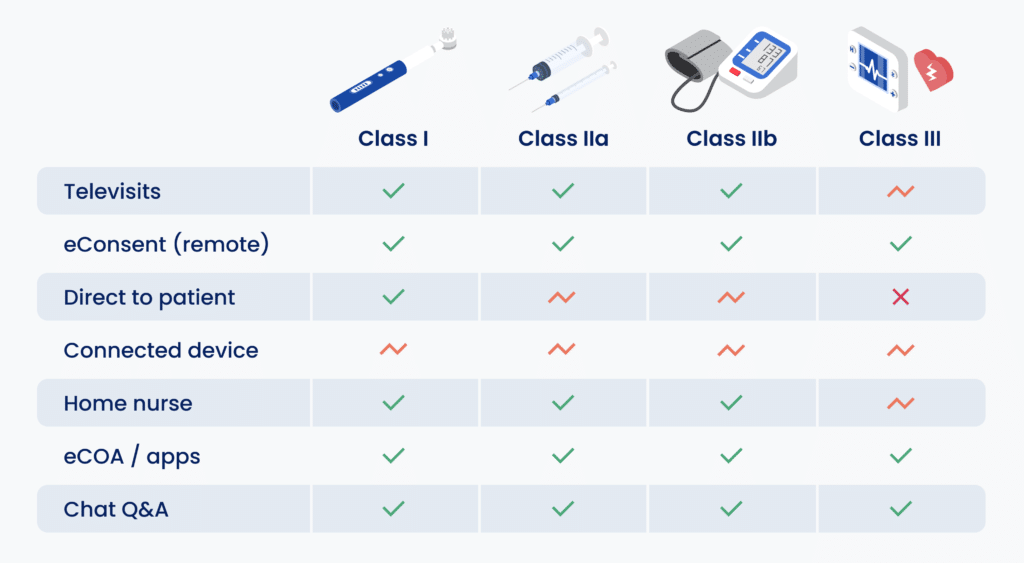
- For Class I to II b devices, it is usually possible to conduct all visits through televisits.
- For Class III devices, the initial implementation or surgery will almost always need to happen on-site, but it is often possible to do some follow-up visits remotely.
eConsent
eConsent allows participants to enroll for trials from the comfort of their homes. A trial’s protocol may include online questionnaires (via web, mobile or native app) to capture everything from eligibility criteria to screening questions to eSignature.
- eConsent can be incorporated into the protocol for any device classification as long as IRB/ethics board requirements are met.
- If a study protocol calls for informed consent to be collected in-person (e.g. directly prior to the implantation of a class III device), other aspects of enrollment such as eligibility and screening questionnaires can still be done remotely.
In Europe, the hurdle is quite high for a manufacturer to get direct access to a patient to collect medical device data due to the General Data Protection Regulation (GDPR). If a lay user device can be found at the local drugstore, then it may be possible to go directly to the patient as long as the legal requirements for GDPR, Health Insurance Portability and Accountability Act (HIPAA) and Good Clinical Practice (GCP) are being followed.
It also may be possible to obtain explicit consent from patients willing to be contacted by the manufacturer for further research, however, it’s important to fully understand the ethical requirements involved in that possibility. In such a case, the patient must be kept informed of changes and the manufacturer must be as transparent as possible.
Direct-to-patient
Where permissible, medical devices can be shipped directly to patients.
- Class I devices can almost always be sent direct-to-patient.
- Class IIa and sometimes Class II b—depending on the particulars of the device—can often be shipped directly to participants.
- Class III devices typically cannot be shipped to a participant as they require clinical supervision or implantation (i.e. surgery).
Connected Devices
Connected devices can be shipped to patients, and can then automatically monitor and collect data and transmit it to study sites or, ideally, push it directly to the trial database. These can be used to monitor activity levels, step count, pulse-oximetry, and more.
- Class III devices such as a pacemaker, deep brain stimulator, or insulin pump require implantation and setup by a clinician on-site, but the subsequent monitoring can be done virtually.
Bluetooth is changing the game for medical devices—and not just for patients. Bluetooth-enabled stethoscopes can now transmit a patient’s heartbeat into paired wireless headphones (or even hearing aids) worn by a physician. Similar remote technologies are gaining popularity as televisits become part of the new normal. Of course, all these new medical devices require extensive testing and require regulatory approval.
Home health nursing
Home health nurses can be enlisted by researchers to travel to collect specimens and provide nursing care in a patient’s home, thus reducing or even eliminating the need for participants to travel. Depending on the type of device or data to be collected, there may be several activities that require the professional oversight and skills of a home health nurse.
- Class I to IIb devices trials can easily enlist the help of a home nurse, as required.
- Class III devices may require some on-site nursing care, although a hybrid approach is possible in many cases.
ePRO and eCOA
Electronic Patient-Reported Outcomes (ePRO) and Electronic Clinical Outcome Assessment (eCOA) offer questionnaires online or via an app instead of on paper. Importantly, with the right platform ePROs can be scheduled or triggered by advanced rules or automation workflows, eliminating the need to contact participants individually.
- Class I to IIb devices trials can easily enlist the help of a home nurse, as required.
- Class III devices may require some on-site nursing care, although a hybrid approach is possible in many cases.
Chat and video calling
Video brings human connection into a clinical trial in many ways. For example, an investigator from a centralized site can talk a participant through the consent process via video call. Follow-ups can be done online through video conferencing. It can also be used to assess if patients are using a device correctly: Is the blood pressure cuff on properly? Are the body sensors in the right spots?
- All classifications of medical devices can benefit from chat and video calling as part of their study protocol, reducing the need for site visits where appropriate.
DCT offers a myriad of benefits, from lowered admin workloads to higher participant retention. By strategically leveraging a whole host of remote tactics—televisits, eConsent, connected devices, ePRO, direct delivery of medical devices to participants’ homes, video calling—medical device trials can streamline their study protocols. Best of all, these can be applied to a wide range of medical device studies.