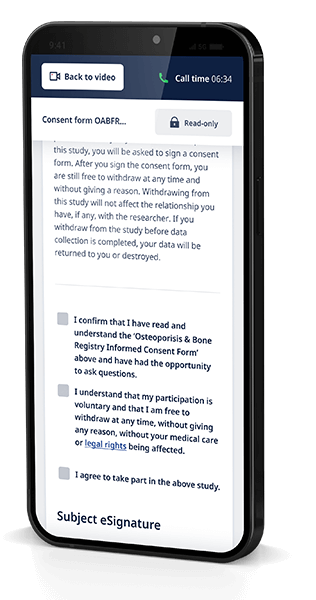
 The informed consent process for clinical trials is often misconstrued as a single event—a signature hastily scribbled on a piece of paper. This is far from what is required by the ethical committees that work alongside regulatory bodies, like institutional review boards (IRBs). Instead, IRBs outline an ongoing and involved process that ensures participants know exactly what they are getting into. Electronic consent (eConsent) platforms advance the consent process by making content more comprehensive, accessible, and digestible for participants.
The informed consent process for clinical trials is often misconstrued as a single event—a signature hastily scribbled on a piece of paper. This is far from what is required by the ethical committees that work alongside regulatory bodies, like institutional review boards (IRBs). Instead, IRBs outline an ongoing and involved process that ensures participants know exactly what they are getting into. Electronic consent (eConsent) platforms advance the consent process by making content more comprehensive, accessible, and digestible for participants.
Here we’ll cover how eConsent benefits trials and aligns with IRB ethical commitments, how to prepare your IRB submission for approval, and the future impact of large language models on the informed consent process.
Informed consent is more than just a signature
Ethical committees are the administrative bodies that protect research participants’ rights, privacy, and welfare. They review informed consent processes to ensure patients understand the benefits and risks of participating in a clinical trial. In the US, these FDA-mandated groups are called IRBs; elsewhere, they are known as independent ethics committees, ethical review boards, or research ethics boards.
Informed consent is an important component of an IRB submission but is more than just a signature.
The FDA defines informed consent in three elements:
- Providing a patient with all the information they need to make an informed decision
- Allowing opportunities to test a patient’s understanding and for patients to ask questions
- Continuing patient updates about impacts on their safety and wellbeing
In other words, informed consent must be holistic, interactive, and ongoing.
eConsent is more than just digital paper
The FDA encourages using eConsent because it provides additional opportunities for patients to connect with trial material and to stay updated throughout the process.
When properly designed, eConsent provides participants with alternative ways to view and process informed consent material outside a clinical setting, new possibilities for assessing knowledge and asking questions, options for participants to stay updated on pertinent information, and a cleanly-tiered presentation of information to satisfy IRBs without confusing participants.
Benefits of choosing eConsent
In addition to the consent process, eConsent platforms often include recruitment, screening, enrollment, and trial onboarding tools. Unlike paper forms, researchers will not have to re-enter data collected by eConsent into the trial database before use.
And the benefits of using eConsent for a trial go beyond saving trees and time—eConsent can vastly improve recruitment and retainment efforts and streamline data flow.
Real-life example: COVID-RED
For example, Castor’s eConsent tool helped Julius Clinical recruit over 17,825 participants in just 15 weeksfor their COVID-RED (Remote Early Detection) study COVID-RED provided participants with a wearable medical device and app that alerted them about a possible COVID-19 infection in real-time, so they could get tested before symptoms appeared. Travel restrictions and a high-risk subset of participants made onsite informed consent impossible.
Participants completed the enrollment process in the safety of their homes using Castor’s eConsent tool. eConsent data went directly to Castor’s electronic data capture (EDC) system so that participants could receive their wearables and get feedback as soon as possible.
This trial would not have been possible during a global pandemic without eConsent.
How to include eConsent in your IRB submission
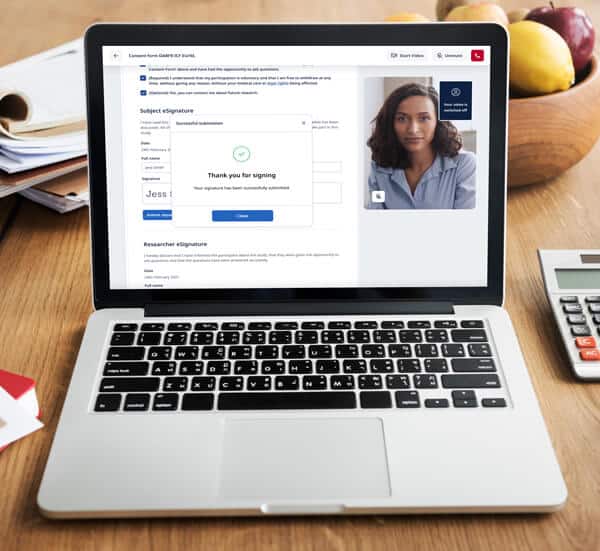 If you plan on including eConsent at any point in your clinical trial, you must submit your plans to your IRB for approval. The IRB will want to see clear documentation detailing how eConsent supports participants’ decision-making during the consent process and throughout the trial. Developing your eConsent before submitting results in fewer requests for changes by the IRB—saving you time and money.
If you plan on including eConsent at any point in your clinical trial, you must submit your plans to your IRB for approval. The IRB will want to see clear documentation detailing how eConsent supports participants’ decision-making during the consent process and throughout the trial. Developing your eConsent before submitting results in fewer requests for changes by the IRB—saving you time and money.
IRB members must engage with the same materials as participants to understand their effect. So they will need access to all the electronic material used in the eConsent tool, not just the content. They will consider interface usability, whether the material is accurate and suitable for participants, and the validity of hyperlinked information. Although IRB rules vary by locale, here are a few elements to consider as you prepare for submission:
Element #1: Consent workflow
Include whether the consent discussion will be face-to-face, remote, or a combination. If not face-to-face, the IRB will review whether the procedures effectively meet the goals of informed consent.
Questions to consider:
- How will participants provide proof of identification?
- How will participants be approached for consent? When will the question be asked?
- What platform will host the informed consent discussion (for example, smartphone, video calls, Castor eConsent videoconferencing)?
Element #2: Patient comprehension assessment
 An IRB submission must clearly explain how researchers will ensure patients meet the criteria of informed consent.
An IRB submission must clearly explain how researchers will ensure patients meet the criteria of informed consent.
Consider:
- How will the study be explained to participants?
- How will their understanding be assessed (for example, popup quizzes)?
- How will questions be answered?
Element #3: eSignature process
Although an electronic signature (eSignature) is an integral aspect of eConsent, eSignature requirements vary by country, impacting global eConsent adoption. For example, Germany accepts both eConsent documents and eSignatures, while its neighbor France is wary of eConsent and does not allow eSignatures.
In the US, a valid eSignature can be the subject’s typed name or even a checkmark, as long as the symbol is logically associated with the person making it. Some software solutions, such as Castor’s eConsent, incorporate a valid eSignature in their platform.
Consider the following:
- How will the electronic signature be created?
- How can the signature be proven legitimate?
According to HIPAA regulatory requirements, eSignatures are only valid if participants can get an email or paper copy of the contract. In your application, consider how participants can get a copy of their consent (ex: download a pdf, receive an emailed form, or receive a paper form in the mail).
Element #4: Special circumstances
Finally, an IRB submission may need a section to address special circumstances related to eConsent per a trial’s unique needs.
The section might answer questions such as:
- How will assent and parental permissions be obtained for minors?
- Do participants need to be reconsented during the study?
- Will caregivers take part in the study alongside participants?
- How will the translation be addressed for non-English speakers? How will consent be witnessed in this scenario?
- What if a participant doesn’t have the right technology to use eConsent?
Large language models and eConsent
Large language models (LLMs) hold exciting opportunities to simplify the interactive elements of informed consent for patients and improve workflow for investigators and IRBs.
For example, researchers can use LLM-generated video content that has passed human approval to engage participants in the informed consent process. Also, incorporating LLM chatbots in the eConsent process could answer patients’ questions in real time. Chatbots thoroughly trained in the trial protocol could replicate the onsite experience at home, reducing patient travel requirements and site burden.
The research community isn’t ready yet to incorporate patient-facing tools powered by LLMs, like chatbots, into the consent process. But LLMs hold the immediate potential to lighten the workload for trial staff and IRBs by generating content for human review and streamlining processes.
eConsent holds enormous potential for making informed consent more accessible for trial participants and simpler for sponsors and investigators. It also aligns with IRB efforts to ensure participants understand exactly what they are getting into when signing up for a trial. When preparing your submission, consider how the elements unique to eConsent, such as eSignatures and content delivery, will fit IRB requirements.