In June 2011, pharmaceutical behemoth Pfizer announced their plans to secure online participant consent using video/multimedia in an upcoming study. The goal was to enroll about 600 participants from ten states across the United States to assess the safety and efficacy of Detrol LA (tolterodine tartrate), a treatment for overactive bladder. The announcement marked a major first in clinical trials, offering hope of accelerated clinical trials while improving quality and diversifying the participant base.
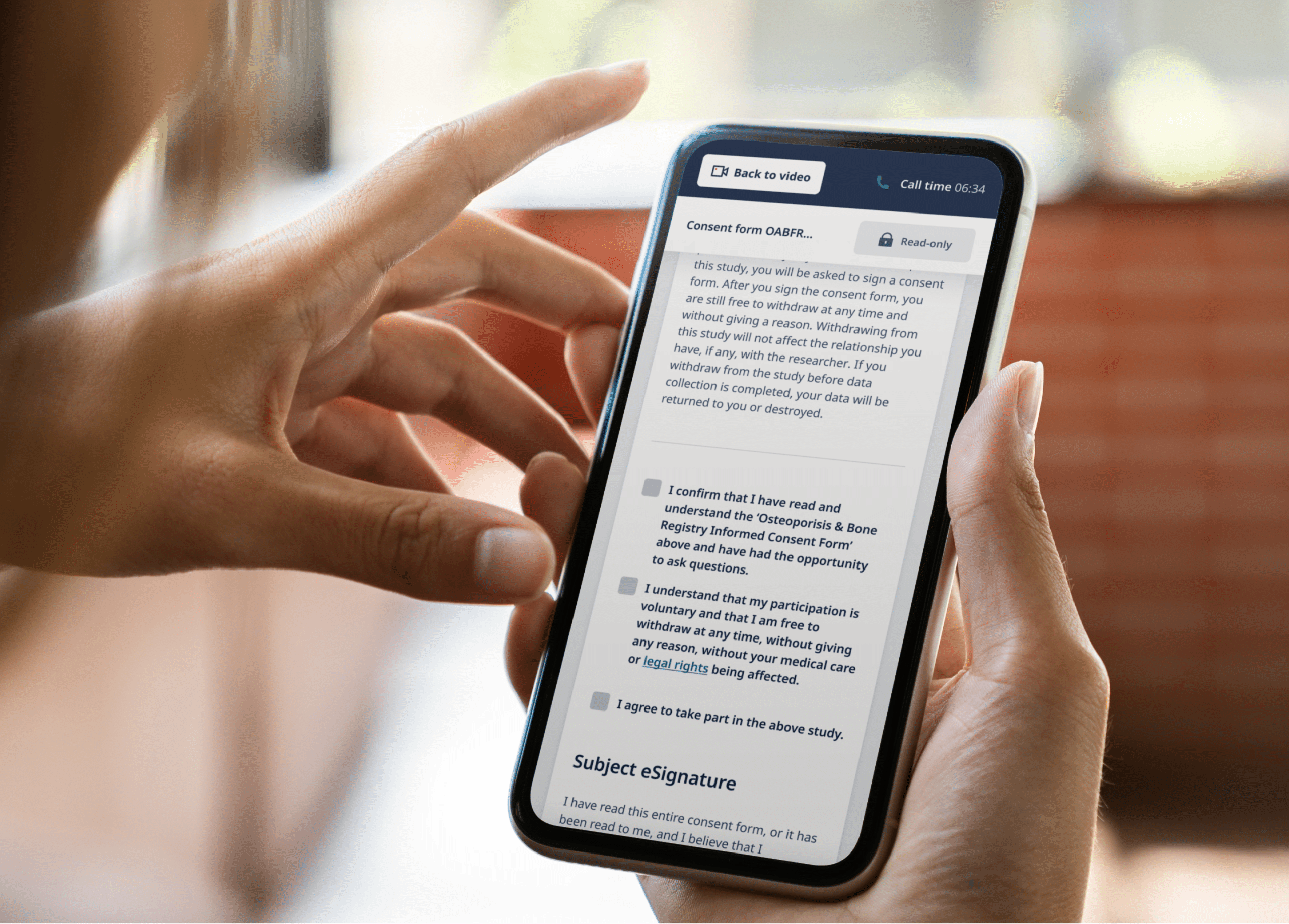
Ultimately, the clinical trial was halted after failing to recruit a sufficient number of participants. Its downfall was attributed to trying out too many ideas at once, ranging from online recruitment and consent to at-home study drug delivery and remote tools for all data reporting. Still, Pfizer noted valuable takeaways and vowed to refine their strategy and try again. Most importantly, this study represented the first foray into eConsent.
Progress in eConsent solutions over the next decade was slow, with the clinical research industry overdue to implement quick, secure, and flexible enrollment and consent options. Then the COVID-19 public health crisis arrived and quickly became a major catalyst for eConsent adoption. The COVID-19 pandemic accelerated the rollout of decentralized trials (DCT) and hybrid trials are rapidly becoming part of the new normal so eConsent is becoming ever more critical in supporting clinical research. So far, evidence indicates that the use of eConsent has advantages over traditional paper methods by:
- Offering better participant experience
- Reducing errors
- Improving efficiency
- Eliminating administrative overhead
- Improving document management
- Supporting better version control
- Improving remote monitoring
What is eConsent?
In brief, eConsent is the digital version of informed consent—and one of the first interactions participants have with a study. It must meet all the criteria of informed consent, simply removing the paper from the site and instead using a tablet or computer to capture consent instead. Remote eConsent represents a paradigm shift, where consent can be obtained remotely in the comfort of a participant’s home, local clinic, or anywhere else convenient. It allows participants to be screened, give consent, and enroll into trials remotely without the burden of visiting a research site.
As eConsent platforms mature, exciting new interactive and visual features are being added to increase participant engagement and comprehension, such as:
- Audio recordings
- Images & Videos
- Video Calling
- Screen readers
However, even robust engagement materials are not meant to replace the important discussion between the participant and site staff. The clinical research site will continue to play a critical role in the consenting process.
The evolution of eConsent
Since it was first instituted in the 1960s, written consent to participate in research studies has undergone very little change other than tightening of regulations and increased participant education of their rights. At that time, it was decided that for research to be considered ethical, participation must be done in a voluntary and informed manner. This decision resulted in an elaborate, paper-based consent process used in all studies until recently. Unfortunately, the goal of informed consent was not always met: to explain research to study volunteers so they could offer truly informed consent.
In March 2015, however, the FDA signalled a seismic shift when it proposed its first ever guidelines for implementing eConsent and laid out an electronic consent meaning. The adoption of eConsent was slow, however, due to concerns of potential risks, longer timelines, and higher costs. There was an overall hesitancy from clinical researchers to embrace an entirely new approach to consent. As is true in all technology adoption life cycles, crossing the chasm to widespread adoption takes time.
eConsent has increasingly become more accepted over the years, however it wasn’t until the COVID-19 pandemic began that it was embraced on a worldwide scale as never before. As research centers closed down, teams working on in-progress studies needed a method to remotely re-consent participants due to protocol amendments. Previous objections to adopting remote consent were overcome by necessity. The industry is now seeking remote consent as a standard for trials instead of novel functionality—and they’ll have plenty of fresh evidence and case studies to back up their requests.
What are the Benefits of eConsent?
eConsent benefits are numerous, offering advantages to participants, sponsors, and site staff. Such eConsent advantages include quality data collection, improved productivity, automation of reporting, increased valid consent, and improved participant access and engagement.
1. Supports quality data collection
eConsent supports data quality by standardizing the consent process, automating versioning, and reducing ICF entry errors. Depending on the platform, eConsent can also use embedded HIPAA-compliant authorization forms, thereby ensuring FDA compliance and necessary documentation at every step. In the EU, the GDPR’s most recent guidance on eConsent requires “an effective audit trail of how and when consent was given, so you can provide evidence if challenged” and “an appropriate cryptographic hash function to support data integrity.” Fortunately, some eConsent solutions offer access control, encryption, and traceability.
Quickly identifying and correcting errors is also on the list of eConsent benefits. It can trigger timely re-consent notifications linked to protocol amendments or safety updates, ensuring that a study is not stalled or stopped. It also reduces transcription errors since data fields (eg. date of consent) can be copied automatically into the electronic data capture (EDC) system.
2. Improves productivity
Another benefit of eConsent is increased productivity in decentralized or hybrid trials—which is great for both a sponsor’s budget and timeline. Site coordinators spend a significant amount of time hosting onsite source monitoring with CRAs from multiple trial sponsors. Managing paper ICFs is burdensome and error-prone for these inspections, not to mention the required storage, tracking, and maintenance of records. But with eConsent, CRAs can check data remotely without visiting the site, allowing them to concentrate on other site duties (e.g. safety monitoring). Or, a central team can review data in the core system and case report forms (CRFs) for all sites, further increasing team efficiency.
3. Allows automated reporting of data
Sponsors need to know where sites and participants are in the enrollment process to identify delays and potential risks. The automated data reporting built into some eConsent programs addresses this issue effectively. For example, it can run automated edit checks on completeness of ICFs and trigger alert messages to sponsors in case of eConsent errors. It can also leverage interdependency with core systems such as interactive voice response system (IVRS), CRFs, or the clinical trial management system (CTMS). All of these features can catch flaws in the intake, enrollment, and continued consent process before they threaten data in the study.
4. Increases valid consent
Every participant in a study represents an investment of time and money—and possibly the key to meeting required participation levels. Invalid consents, however, require hard-earned data to be tossed out. Flaws in the intake and enrollment process can create errors in the order in which consent is documented. At times, sites move along in the trial process without getting the ICF first— thus invalidating data in the study. eConsent, however, requires initials, signatures, and checkboxes coupled with automatic date and time stamps on documents, so trial materials are always audit-ready. eConsent also ensures that participants are always given the most recent IRB-approved version of consent forms, auto-triggering notifications when a re-consent is necessary.
Another way an eConsent program supports valid consent is by communicating clinical trial information in easy-to-understand formats. Through the use of interactive multimedia components such as videos, images, and audio, eConsent can make it much easier for participants to fully understand the information being presented to them. With better comprehension, participants are empowered to make decisions that they feel confident about.
5. Improves participant access
A well-documented problem plaguing research trials is a lack of diversity and inclusivity. In the past, participants were often pulled from populations living near study sites, compounding the problem of underrepresentation. With increased awareness, more trials are actively seeking out diversity of race, gender, and geography amongst participants so that the studied population is representative of the distribution of the disease.
Individuals who have high-risk health conditions or who live in rural areas are much more likely to be able to participate when eConsent is used because it eliminates travel. eConsent can easily be adapted to meet the needs of a wide variety of participants by addressing language or literacy barriers, hearing or vision impairment, and other special needs. For example, the use of audio recordings, adjustable font sizes, and multiple language formats easily serve the needs of a wider audience.
eConsent also makes it easier to accommodate participant needs. For example, sites can observe which form sections a participant is spending more time on. If participants have questions, these can be quickly resolved through video chats with the site team. This allows site staff to streamline services and allocate resources according to real needs instead of projected ones.
6. Increased patient engagement & retention
eConsent can ease many common anxieties that participants often experience during the consent process. By enabling a more interactive experience, eConsent allows participants to go through information at their own pace. When signing a document in person, participants may feel pressured and anxious as they review and sign the document. eConsent offers a refreshing alternative, where participants can take their time with each section of the document, even pausing to reflect or ask for input from family and friends. Any questions that come up can also quickly be answered by a researcher through the video chat feature. Because participants feel actively involved in the study process, they are more likely to feel invested and engaged with the study—leading to higher participant retention.
The eConsent process: from recruitment to retention
A participant’s journey through the informed consent process does not begin and end with a signature. It spans across recruitment, screening, enrollment, consent, and retention. To expand participant access to trials and democratize clinical research, a streamlined and remote-enabled participant experience along all of these steps is critical—and the right eConsent solution will help to do just that.
Modern researchers are tapping into the power of social media to reach a broader audience. But once they’ve sparked the interest of a potential participant, the next challenge is to convert that interest into engagement. To do so, an online screening questionnaire can determine eligibility to participate in the study. Depending on the eConsent platform used, potential participants may receive an email directing them to a customized enrollment page to share trial information and answer questions.
Importantly, digital participant enrollment can make the difference between hitting or missing your recruitment targets. An eConsent enrollment page can use multimedia components (eg. videos and audio recordings) to break down complicated trial information and help participants understand what a trial aims to achieve, and the risks and responsibilities of the participant. Where formerly participants may have been confronted with a 30-page enrollment form laced with legal jargon, with eConsent a thorough online presentation can address participant concerns, empower them, and strengthen their enthusiasm and commitment.
Video conferencing allows a clinical researcher to join a participant online and answer questions directly, building further trust and commitment to a study. This also allows the study team to verify the identity of the participant.
Finally, participant retention is one of the most important aspects in ensuring that a clinical trial continues to move forward. Unfortunately, it is also often one of the biggest obstacles. An eConsent solution, however, can alert site staff to follow up with participants who are falling through the cracks by missing check-ins, not entering in data, or skipping lab visits. It also ensures that critical trial information is always available online, further helping with retention as participants can re-read materials or watch videos again.
Meeting the complexities of informed consent
Clinical researchers are required to fully explain details of treatment to potential participants. It’s important that participants fully comprehend the risks and benefits before deciding whether or not they wish to consent to it. This process is known as informed consent. (Read about it in-depth here: Understanding the Nuances of Consent in Clinical Research.)
What is the criteria of valid informed consent?
Disclosure of information. participants have the right to autonomy, or the right to make decisions about their own health and medical treatments. Researchers must disclose enough information about the trial so that the participant can make an informed decision. Such information could include what the treatment involves, the possible benefits, and risks as well as the likelihood of any risks occurring.
Competency of the participant (or surrogate) to make a decision. This refers to a participant’s decision-making capacity. The participant must have the ability to make and be held accountable for their own decisions. participants must be able to evaluate the personal benefits and costs of each option and see how they relate to their own values and priorities.
Voluntary nature of the decision. participants must be free to consent to or refuse treatment and voluntarily give consent without any coercion or duress. |
eConsent and Regulatory Compliance
Regulations pertaining to the use of eConsent vary amongst countries. At the time of writing, no EU regulation or guidance about eConsent in clinical trials exists.
In general, the Institutional Review Board (IRB) or ethics committee requirements for paper and electronic consent are the same, although countries and regions differ in exact policy, so an understanding of local IRB guidelines is essential. When making a submission for an eConsent-based study, it’s important to address the following questions:
- Who will conduct this discussion?
- How will participants be identified?
- How will participants be approached for consent?
- When will participants be asked to provide consent?
- What video conferencing platform will be used for remote consent discussion? (eg. phone, Skype, Castor eConsent, etc.)
Using eConsent in a country that does not allow electronic signature
In countries that do not accept eSignature, it may be possible to set up an alternative workflow. Check with your local IRB if a participant can sign a paper form while on a video call, and then mail in that form. Although this method entails paperwork, you retain two key benefits: the eClinical platform tracks the consent status, and participants can access trial information online at any time.
The future of trials: eConsent
Studies show that on average, the cost of developing a drug is $2.6 billion, and for every day a trial is delayed, sponsors lose a whopping $600,000 to $8 million. eConsent removes several common sources of delays while boosting enrollment and retention. For in-progress trials, eConsent supports efficiency in data processing and continuous regulatory compliance.
The eConsent model can be a flexible, user-friendly, and secure solution to informed consent when built and used appropriately. It can benefit study participants by putting them at the center of the consent process and offering unparalleled patient education through multimedia elements. It can also offer efficiency gains to researchers by simplifying enrollment and increasing retention. Long after social distancing ends, remote eConsent functionality will no doubt continue to address the growing adoption of decentralized and hybrid trial designs.
Understandably, some sponsors are hesitant to make the leap to eConsent due to the uncertain regulations or fear of extended startup times and expenses. Switching SOPs and workflows once built for the paper and site process will require change management. However the benefits are massive and—as with the smart phones we carry—in a few years we may wonder how we ever got by without remote eConsent solutions.
Castor’s remote eConsent solution is a powerful platform that puts the participant at the center of the consent process and reduces workload for site staff. Reach out to one of our friendly Castorians to find out how we can set up your next study for success.