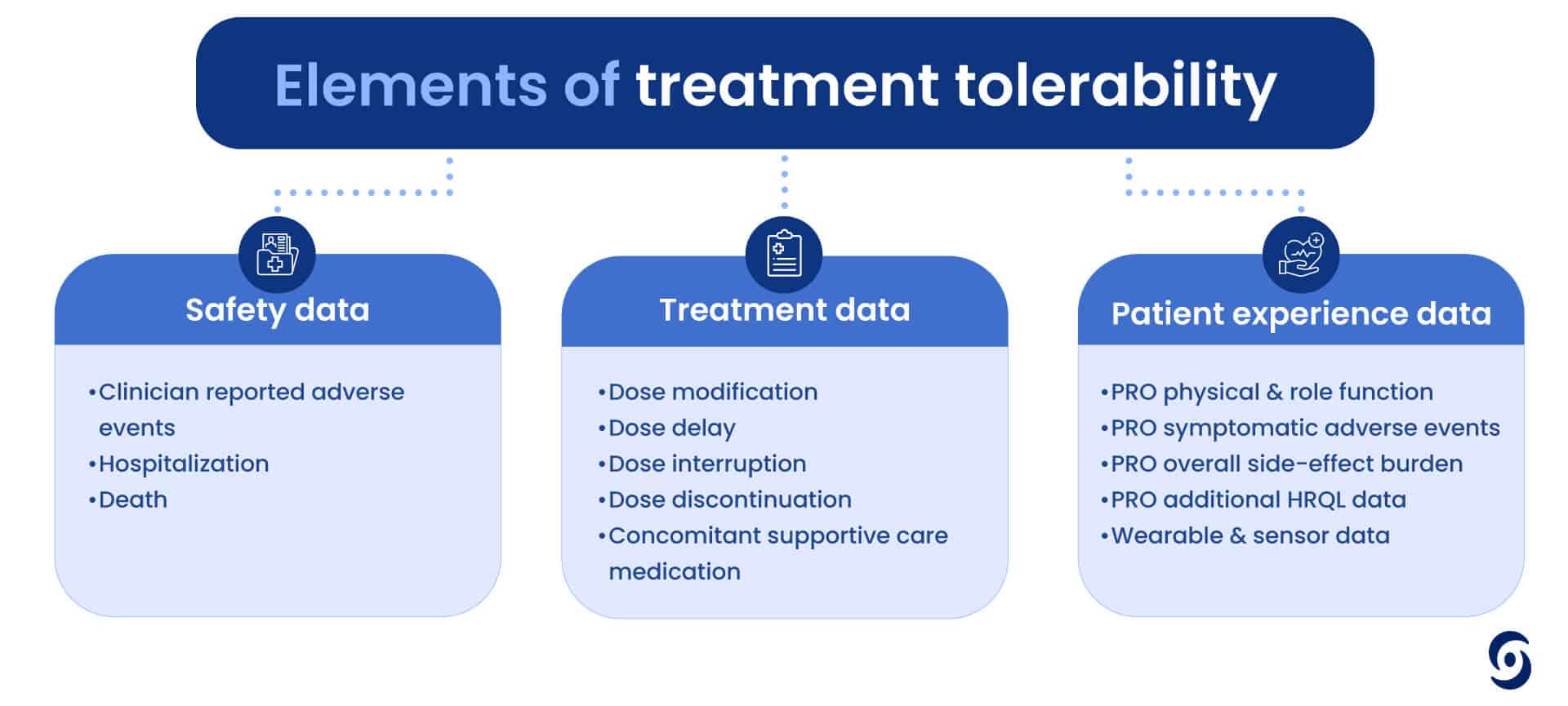
A new BP monitoring device by Aktiia, promises to reinvent the practice of patient monitoring by offering continuous, cuff-less, and non-invasive blood pressure tracking. During a recent discussion between Castor’s CEO, Derk Arts, and Jay Shah, Chief Medical Officer at Aktiia, they explored the transformative journey of Aktiia’s innovative device and the partnership with Castor, which has been crucial for capturing data essential for regulatory milestones.
The Genesis of Aktiia’s Breakthrough Device
The webinar kicked off with Jay detailing Aktiia’s origins and its path to innovation in blood pressure monitoring. Founded in Switzerland, Aktiia has evolved its technology over two decades, culminating in a device that operates seamlessly within users’ daily lives. This technology not only measures blood pressure continuously but does so with a precision that caters to diverse patient cohorts across multiple therapeutic areas.
Derk then emphasized the critical role of continuous monitoring in combating the global hypertension epidemic. With hypertension affecting billions and contributing to various health complications, Aktiia’s device offers a proactive approach to managing this pervasive condition.
“Continuous blood pressure monitoring could well be the most significant weapon in our arsenal against the global epidemic of hypertension.”
– Derk Arts, CEO of Castor
Bridging Technology with Clinical Efficacy
The conversation delved into how Aktiia’s device integrates advanced photoplethysmography (PPG) technology with user-friendly design. This integration is crucial not just for patient compliance but also for the accuracy and reliability of the data collected. Such data is vital for regulatory approvals and advancing medical research.
Jay Shah outlined the regulatory journey of Aktiia’s device, emphasizing the rigorous scientific and regulatory standards that have been met to ensure the device’s efficacy and safety. The partnership with Castor has been instrumental in streamlining data capture for these regulatory milestones, demonstrating the power of combining innovative devices with robust data management platforms.
“Our device is about more than just monitoring; it’s about understanding patient health in real-time, enabling proactive healthcare interventions.”
– Jay Shah, CMO of Aktiia
The Impact of Real-World Evidence and Patient-Reported Outcomes
As healthcare continues to evolve towards more personalized and patient-centered models, the integration of real-world evidence (RWE) and electronic patient-reported outcomes (ePROs) becomes increasingly important. Aktiia’s technology enables the capture of vast amounts of valid blood pressure data, which, when combined with ePROs, provides a comprehensive view of patient health.
The discussion highlighted how this data could transform clinical research and patient care by providing insights that are not only more accurate but also more reflective of patients’ real-world experiences. This shift is crucial for developing more effective treatment protocols and enhancing patient engagement and compliance.
“Integrating real-world data with patient insights allows us to see the full picture of treatment effects, paving the way for truly personalized medicine.”
– Derk Arts
Conclusion: A Vision for Future Healthcare
In conclusion, the collaboration between Aktiia and Castor is a prime example of how technological innovation can meet clinical needs. The ability to monitor blood pressure continuously and non-invasively represents a significant leap forward in patient care and clinical research.
The future of healthcare lies in leveraging such technologies to reduce burdens on patients and clinicians alike, making healthcare more accessible, efficient, and effective. As we continue to embrace these innovations, the horizon of what’s possible in medicine expands dramatically.
For a deeper dive into the discussion between Derk and Jay, click the button below to watch back on the LinkedIn Live recording.